

This new virus is rapidly spreading globally. In just 1 month the virus has infected people in 23 countries causing a worldwide pandemic. There is no effective medical treatment. However, there is a natural solution that shows much promise. read more

The prescription and over-the-counter medications you may be taking, especially if they are generic, may be doing you more harm than good. And the situation is getting worse. read more

If you use teabags, you are probably consuming a substantial amount of plastic with your tea. read more

Issue 16-5 covers: Is Coconut Oil Toxigenic?—Protect Your Vision with Coconut Oil—Natural Versus Allopathic Medicine—Coconut Oil for A Healthy Planet. read more
Unless you are a hermit you have heard about the coronavirus—a deadly new virus that is rapidly spreading worldwide. The first case was reported in Wuhan, China on December 21, 2019. As of February 10, 2020, mainland China has reported over 40,000 confirmed cases and nearly 1,000 deaths.
On January 21, 2020, the first US case was confirmed— a patient in Washington state who had recently visited Wuhan, China. A second case, in Illinois, was confirmed three days later. This patient had also recently returned from a visit to Wuhan. As of February 8, 2020, there were 12 confirmed cases in the US.
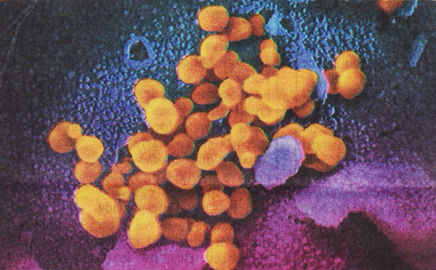
Coronavirus —US National Institutes of Health
The corona virus has also been reported in at least 23 other countries, including Canada, Australia, France, Japan, Thailand, South Korea, Taiwan, Vietnam, Singapore, Saudi Arabia, and Africa. There have been hundreds of cases reported outside of China, and the numbers are rising. In just over one month the virus has spread worldwide and appears to be blooming into a worldwide pandemic. The World Health Organization has declared a global health emergency. The Chinese government has shut down all travel in and out of Wuhan — a city with a population of 11 million — and the surrounding area in an effort to contain the spread of the disease. Travelers returning home from recently visiting China are being quarantined for at least two weeks.
Coronaviruses (CoV) are a family of RNA (ribonucleic acid) viruses. They are called coronaviruses because the virus particle exhibits a characteristic ‘corona’ (crown) of spiked proteins around its lipid envelope. CoV infections are common in animals and humans. Some strains of CoV are zoonotic, meaning they can be transmitted between animals and humans, but many strains are not.
In humans, CoV can cause symptoms range from those resembling the common cold to deadly, such as Middle East Respiratory Syndrome (caused by MERS-CoV), and Severe Acute Respiratory Syndrome (caused by SARS-CoV). Patients infected with the new coronavirus, named 2019-nCoV Acute Respiratory Disease (2019-nCoV), usually exhibit severe coughing, which may be accompanied by labored breathing. All cases show radiographic evidence of pneumonia.
Currently, there is no cure or effective medical intervention to treat the virus. The body must fight the infection on its own. Those with weak immune systems, generally the very young and the elderly or those who are in poor health, are most vulnerable. At some point there may be a vaccine developed for it, but that will be a long way off.
Take heart, you are not helpless against this new virus, there is a natural solution that may prove quite useful—coconut oil. The coronavirus is a lipid coated virus, meaning it is encased in a fatty envelope. The medium chain fatty acids in coconut oil, particularly lauric acid, have been shown to kill lipid coated viruses. Coconut oil consists of 63 percent medium chain fatty acids; nearly 50 percent of the oil is lauric acid.
The antiviral action is attributed to solubilizing the lipid envelope of the virus causing it to disintegrate, killing it. In addition, evidence also indicates that lauric acid may interfere with signal transduction in cell replication, preventing the virus from reproducing. All medium chain fatty acids have some antiviral properties; lauric acid is by far the most potent. It is important to note that while MCT oil is composed of 100 percent medium chain fatty acids, it contains no lauric acid, and therefore, would not provide the best protection against the coronavirus. At this time, the daily consumption of coconut oil, at approximately 3 tablespoons (45 ml) per day, might be your best defense.
Doctors Fabian Dayrit, PhD and Mary Newport, MD have co-authored an article on the coronavirus and how it may be possible to treat it using coconut oil and/or monolaurin. You can read the full article here
The contaminated vitamins caused me severe illness. At the time, I was relatively young and healthy and recovered without problem. However, what if someone who was older, already sick, or suffering from a serious chronic condition took these tainted pills. What would happen? Their response may have been much worse than mine and perhaps even life-threatening. Many people rely on medications to help control chronic conditions and infectious illnesses. What if they got hold of a drug that was contaminated or was below stated potency, what could happen?

In 2011, Betty Hunter, an Arizona resident suffering from lung cancer, went to her oncology clinic to receive a cancer medication called Avastin. She quickly developed chills, vomiting, cramping, and a fever. It was later discovered that Hunter’s doctor had obtained the drug from a Canadian supplier, which had purchased the drug from a Turkish manufacturer. Analysis revealed that the fake medication not only lacked the active pharmaceutical ingredient in Avastin, but contained tap water and mold.

Veronica Diaz was a healthy 22-year-old woman who had mild anemia. She was put on a course of 10 injections of an iron-based medication. During the treatment her health steadily declined. After the seventh injection she become terribly sick and died of liver failure. Investigators determined that she had been given a highly toxic counterfeit. Authorities were unable to determine the source of the counterfeit product due to falsified paperwork.
Severe reactions to pills and medications are often given little notice as they are considered to be simply reactions to the medicines. All drugs have the potential to produce adverse reactions. Generally, these reactions are known to occur in some patients, there is always a risk. You’ve seen the ads for medications with accompanying disclaimer spotlighting some of the many possible side effects. These are the known side effects to drugs that are properly formulated and stored until use. An entirely new set of adverse effects can occur in addition to these, if the drugs are improperly produced and handled—products that may contain contaminants or adulterants, are incorrectly formulated, or otherwise altered. This is becoming a growing problem worldwide.
The Need for Generic Drugs
Drugs have become an essential treatment for many illnesses both chronic and acute. We have come to depend on them. By age 65 over 20 percent of us take an average of five medications and by the age of 85 that number increases to eight. Drugs can be expensive, costing thousands of dollars a year. We need a more economical option. Generic drugs offer a possible solution.
Generic drugs are supposedly bioequivalent to brand-name versions and assumedly provide the same therapeutic effects. Brand-name prescription medications are often extraordinarily expensive. According to the US Food and Drug Administration (FDA), generics cost on average 85 percent less than branded drugs, making them very popular. Ninety percent of the prescriptions filled in the United States are for generics.
Lipitor, a brand-name cholesterol-lowering statin, costs 10 to 50 times more than its generic equivalent atorvastatin. Depending on the pharmacy from which it is purchased, Lipitor costs between $111 and $520 for a 1 month supply of 10 mg tables. In contrast, atorvastatin, the generic version of Lipitor, costs between $10 and $20 for the same amount. In a year, a customer can pay as little as $120 or as much as $6,240 for essentially the same thing. It’s no wonder why generic drugs are a popular alternative.
If an equivalent generic drug can be made for so little, why are brand-name drugs so expensive? Brand-name drugs are patented, generics are not. It often takes many years to develop an effective drug, According to the Tufts Center for the Study of Drug Development the cost to develop and win marketing approval for a new drug is $2.6 billion. Patent protection on drugs usually lasts 20 years from the date of the patent application. Drug manufacturers patent their products and processes early in the research process to protect their intellectual property. However, the time required to demonstrate that the drug is safe and effective and get government approval to sell the product may take several years. Consequently, the practical duration of protection from competition after the drugs finally goes to market is greatly reduced. Therefore, a provision has been made to extend patent protection an additional 5 years to account for the time delay. Generally, new drugs have between 7 to 12 years of protection before the patent expires.
After expiration, the gate is opened for generic competition. Typically, within two years brand-name drugs lose more than half of their sales to generic drugs. For this reason, brand-name drugs are costly in order for the makers to recoup their initial investment and make a worthwhile profit. Since generic drugs are considered to be equivalent to the original, they are assumed to have the same degree of safety and therapeutic value. This allows them to be streamlined into production at relatively minimal cost.
Generics Are Not The Same As Brand-Name Drugs
Both prescription and over-the-counter brand-name drugs have to be tested in clinical trials to prove they are safe and effective before being approved for sale. Generics do not, they just have to show that they produce the same level of the active pharmaceutical ingredient in the blood as the branded drugs. The exact processing methods and procedures, storage and handling conditions, and the types of fillers and other inactive ingredients used can all vary. Manufacturing is expected to follow the FDA’s regulations known as the “good manufacturing practices” or GMP to assure purity and quality of the product.
Generics are loudly touted as a cost savings to consumers and for the most part, they are. We are told that they are “exactly the same as brand-name drugs.” However, this is not exactly true. There are slight differences. The types of inactive ingredients—fillers, binders, coloring, flavoring, preservatives, and such—can vary greatly. The amount and the rate of absorbability of the active ingredient can vary as well. As long as the generic can get within 10 percent above or below the blood concentrations achieved with the branded product, it is acceptable. For instance, one generic may get you a 10 percent lower concentration than the brand and another may get you a concentration that is 10 percent above the brand and, therefore, the two generics can be 20 percent different from each other. Usually, they only vary by 3 to 4 percent one way or the other. With slight variations such as this, it is assumed that most people would not notice a difference.
All drugs are poisons. The blood concentration that distinguishes between a therapeutic and a toxic dose is small. For some drugs, this range is very narrow and even a slight deviation in potency can lead to ineffective or to toxic responses. These types of medications are known as narrow therapeutic index (NTI) drugs. Medications for seizure control, heart arrhythmia, thyroid hormone, blood thinners (warfarin), and lithium are all examples of NTIs. Because the active ingredient in generics can vary from branded drugs and from one generic brand to another, patients using NTIs need to take particular care in switching their medications and consult with their physicians.

Can We Trust Generic Drugs?
Because generics can be significantly cheaper than brand-name drugs some people question if they are of equal quality. Is the inexpensive generic store brand you get from your local grocery store as good as the well-known brand-name sold at the pharmacy?
Although brand-name and generic drugs are said to be bioequivalent, many people who have responded well to a brand do not benefit from generics, and they have no idea why. The generic may cause unexpected adverse reactions or have little or no therapeutic benefit. Because generics are not identical to brand-name drugs, patients’ response may vary. Some people may be more sensitive to the active ingredient than another and experience adverse reactions. Slight variations in potency, even within acceptable limits, may cause adverse reactions. Allergic reactions to inactive ingredients may also influence the effectiveness of the product. For these reasons, you may do well with a branded drug but not so well with a generic, or do fine with one generic but not another. This is why your doctor may change your prescription from time to time. Many doctors recommend that patients start out on brand-name medications to see how they respond and then switch to cheaper generics afterwards, if they are available. If one generic doesn’t work as well as the brand, then another can be tried.
Another reason why some generics may not work well or may cause unusual adverse reactions is due to contamination or adulteration of the product. This is a problem that is far more common than we are led to believe and can have serious consequences. Most of the generics manufactured in the US, Canada, Western Europe, and Israel are as safe and effective as the branded versions, but if they are produced in developing countries your chances of that are significantly lower. The operations in the manufacturing plants in these countries can be a far cry different from those operating in highly regulated US plants.
In countries with poor regulation and oversight any number of problems can arise during manufacturing that could affect the safety and efficiency of the medicines produced. Manufacturers are expected to verify, through testing and monitoring, that the chemicals sourced from various dealers are pure and unadulterated. Testing however, is often ignored, inadequately performed, or even falsified. In order to cut costs and increase profits substitutions can be made somewhere along the manufacturing process, replacing high-quality ingredients with cheaper, and sometimes toxic, ones.
Dangerous Drugs
The chemical diethylene glycol (DEG) is an industrial solvent often used in the production of antifreeze. DEG is chemically similar to glycerin, a harmless inactive ingredient often used in medicines. DEG is less expensive to make and is difficult to distinguish from glycerin without proper testing.
In 2006, barrels of DEG labeled as glycerin were shipped from China to Panama, passing through several brokers in the process, being relabeled at each step with a certificate of analysis (COA) indicating identity and purity of the product. In Panama the DEG was used in the manufacture of 60,000 bottles of cough syrup. When patients began taking the medicine they suffered from a variety of adverse reactions including paralysis and death. It took authorities a month to track down the cause of the problem. In that time, at least 78 people died from the tainted cough syrup.
Even when the substitution of DEG for glycerin was discovered, relabeling by brokers prevented investigators from quickly identifying the source. Each time the fake glycerin changed hands, international brokers created new falsified certificates of analysis, without actually testing the product. The multiple fabricated documents impaired the investigation so that the original perpetuators in China were never held accountable.
This was neither the first nor the last DEG drug related poisoning. In 1937 DEG was used in the manufacture of an antibiotic in the US, resulting in over 100 deaths. Between 1937 and 2008 more than 750 documented deaths in 10 countries have been linked to drugs adulterated with DEG. In 1995, 50 tones of fake glycerin was shipped into the United States, but fortunately was identified before it could be used in manufacturing. In 1997 DEG tainted drugs killed at least 88 children in Haiti, which was traced back to China through multiple brokers and faked documents. Another 33 children died in Guragon, India in 1998 from the same adulterant. All of these deaths could have been prevented if the handlers had properly analyzed and documented the products according to regulations.
Drugs Are Produced All Over the World
Most people don’t think about where their drugs come from or how they are made, their primary concern is to get a product that is affordable. We assume government regulators have screened them all to assure us they are safe.
The number of drug products produced outside the US has increased dramatically since the 1980s. By 2005, the FDA had more drug plants to inspect abroad than it did within US borders. Today, over 40 percent of the finished drugs used by US patients are manufactured abroad, and 80 percent of active ingredients and bulk chemicals used in US drugs come from foreign countries. [1][2] The United States has increasingly relied on drug manufacturing in developing countries—primarily China and India.
Of course, overseas manufacturing can work perfectly well and the FDA contends that it has a reliable review system for all approved drugs. The FDA and its counterpart agencies worldwide monitor the quality and safety of drug and device manufacturing by inspecting plants and validating compliance with their good manufacturing practices (GMP), which are regulations that that describe the methods, equipment, facilities, and controls required for producing safe products. Because it is impossible to analyze every single pharmaceutical item produced, adherence to manufacturing quality standards is the most efficient way for the FDA can ensure drug quality and safety. While noncompliance with manufacturing quality standards does not necessarily mean that a drug is unsafe, it increases that risk by weakening the safeguards.
Manufacturing quality standards for drugs are set by the FDA and by other regulatory agencies cooperatively in guidance through the International Conference on Harmonisation of Technical Requirements of Pharmaceuticals for Human Use (ICH). The ICH brings together the regulatory authorities of the United States, Europe, and Japan to discuss the scientific and technical aspects of product registration. The ICH guidelines are used by the FDA and European agencies in regulating drug quality. All drugs marketed in the United States, Europe, and Japan, regardless of their country of origin, must comply with these regulations.
Before foreign drug manufacturers can market a drug in the United States, they must submit an application to the FDA giving specifics about the drug. If the application is approved, the drug can be marketed in the United States with the stipulation that the FDA can periodically inspect the company’s manufacturing facilities to assure they are complying with GMP.
The FDA requires drug manufacturers to perform testing on their products to ensure the identity, strength, and purity of their products. Under GMP, testing must be performed on incoming drug ingredients and in-process materials as well as finished drugs. Companies seeking to market medicines in the United States must specify the analytical methods necessary to ensure that the drugs and their ingredients meet established levels in their applications to the FDA. The FDA can ensure that a drug meets established specifications by enforcing adherence to the GMP and the specifications contained on the approved drug applications. An important function of testing is to screen for possible harmful contaminants and assure appropriate levels of the active pharmacological ingredients. Failure to comply to proper testing procedures can result in drug recalls and bans.
Prescription and over-the-counter medications and ingredients originate in factories all over the world. They move into the American and European marketplace through supply chains that can involve numerous processing plants, manufacturers, suppliers, brokers, packagers, and distributors. Tons of tainted or substandard drugs pass through these supply chains annually putting millions of people at risk. While laws and regulations are established to reduce the risk, careless handling and fraud allow faulty drugs to end up in any country.
Domestic facilities are typically inspected by the FDA every two to three years. Foreign processing plants are usually inspected once every nine to 11 years. Due to the complex logistics involved and the lack of governmental authority in regulating manufacturing facilities overseas, foreign processing plants receive far less scrutiny from the FDA than domestic operations. Because of this weakness in regulatory control, counterfeit and substandard medicines are able to slip into the country and be sold at pharmacies and be used in hospitals and medical clinics. Such drugs not only rob customers financially but can have grave health consequences.

Counterfeit and Substandard Drugs
Counterfeit drugs are those that may be of poor quality or even useless but are packaged and labeled to appear to be brand-name or generic products. They are produced without any oversight. Their quality is unpredictable and may contain the wrong amount of active ingredients or no active ingredients, as well as potentially harmful contaminants. In all cases, counterfeit medicines are manufactured in clandestine laboratories with little regard to purity and quality. They are essentially fake drugs.
Substandard medications, on the other hand, are legitimately marketed but contain adulterants or improper amounts of active ingredients, or were compromised by improper manufacturing, handling, or storage. Whether the result of deliberate or unintentional acts, counterfeit and substandard products have the potential to cause serious harm and to deny patients the therapy they need.
Counterfeit and substandard drugs encompass all types of products including anticancer medicines, antibiotics, insulin, pain relievers, antihistamines, diuretics, hormones, steroids, blood glucose test strips, medicines for hypertension and cholesterol-lowering, the list can go on and on. Patients depend on these medications. The absence or insufficient amount of the active ingredients in these products or the presence of potentially harmful adulterants can lead to a patient’s poor response to treatment and even to death.
The World Health Organization states that the quantity of counterfeit drugs sold in most industrialized countries with effective regulatory systems and market control, such as the USA, Western Europe, Australia, Canada, Japan, and New Zealand, have less than 1 percent of the market value. While this may not sound like much, the numbers are huge. For prescription drugs alone, 1 percent of the 4 billion prescriptions that Americans use each year would equate to 40 million dispensed fake medications. In addition, hundreds of millions of legitimate, but substandard, generic prescription and over-the-counter medications are sold each year.
Counterfeit and substandard drugs are most prevalent in underdeveloped countries with weak regulatory enforcement and standards. The World Health Organization’s International Medical Products Anti-Counterfeiting Taskforce estimates that fake and substandard drugs account for more than 30 percent of the market in parts of Africa, Asia, and the Middle East. [3] Drugs that cannot pass standards for entry into the US and Europe are not discarded by their makers but shipped to poorly regulated markets. In Africa, which companies consider the safest place to send faulty drugs, doctors regularly find that drugs for AIDS, bacterial infections, and other conditions are underpowered. It is common for doctors there to give patients up to 10 times the recommended dosage of a generic antibiotic in order to have any effect. Some drugs have no effect regardless of the dosage. Those people living in these countries that can afford it, opt for brand-name drugs whenever possible.
Buying Drugs Online is Risky Business
The riskiest place to purchase drugs is online. A team of researchers from the Harvard-affiliated Brigham & Women’s Hospital in Boston, led by R. Preston Mason, PhD, investigated the quality of generic Lipitor available online to US customers. The investigators analyzed 36 samples of atorvastratin obtained from sources outside the US that were manufactured by more than two dozen generic drug companies. After analyzing the chemical composition they were stunned by their findings; 33 of the 36 samples contained impurities high enough to render them useless. [4] Even samples obtained from Canada, but sourced from India, contained impurities. Interestingly, samples made by the same manufacturer but sold in different countries contained widely different impurity levels, evidence that some generic drug companies segment their markets based on quality, sending substandard medicine to poorly regulated markets, such as in Africa, and higher quality drugs to better regulated markets.
Many people shop online to get the best prices on generic drugs. Unless you know and trust your source, this may not be a good idea. Medicines purchased over the Internet from sites that conceal their physical address are counterfeit in over 50 percent of cases. Even medications purchased online from countries, such as Canada, with strict drug regulations are not necessarily safe.
There is no system in place to assure the safety and quality of medicine that was not intended for the US market even if is shipped to the US from Canada. Canadian regulators focus on—and are only resourced to regulate—drugs dispensed to Canadians, not on drugs that are exported to the US.
FDA Inspections
Despite the strict regulations, a huge volume of substandard drugs enter North America and Europe every year. We assume government agencies, such as the FDA, are on a constant vigil, inspecting products and analyzing samples, to assure our safety. But the task is too large and complex for the resources available to police it completely. The FDA regulates many thousands of drugs and medical devices intended for the US market. Millions of tons of medicines that are made at sites around the world are shipped into the country every year.
Within the United States, FDA investigators typically show up unannounced to inspect production and testing rooms and examine records. But overseas, to maintain good relations with the various counties, the FDA announces in advance the vast majority of its inspections, sometimes months in advance. With advanced notice, the companies are expected to participate in scheduling the trip and make arrangements for travel and lodging. Plant officials often plan receptions and special presentations and provide tours of their facilities, taking up the inspectors’ limited time and controlling what the agents see and do. Entertainment, golf trips, sightseeing, upgraded hotel rooms (without cost), and other perks are often added to make the visit amenable. In effect, the company acts as a travel agent and the inspectors become guests of the plant officials. Other than observing the manufacturing process and equipment that they are allowed to see, the products are not closely examined or analyzed. The safety, purity, and effectiveness of the products and compliance to the FDAs good manufacturing practices are determined by looking at company records, which could easily be altered or falsified. In countries where the inspectors do not speak the native language, they cannot read the documents and don’t even bother to examine them. This entire process leaves a lot of opportunity for fraud and carelessness to escape the inspectors’ detection.
The FDA has defended the practice of giving advance notice as the best way to ease the complex logistics of getting visas and ensuring access to the plants. But the resulting inspections are largely “staged,” say a number of FDA staff members. With advance notice, the plants can make anything look like anything. “You give them a weekend, they’ll put up a building,” as one FDA investigator put it. [5] Some companies construct buildings that are kept in pristine condition solely for inspectors, while the actual manufacturing occurs in warehouses and labs elsewhere.
Because of the high demand for inexpensive drugs in the US, the FDA has been lax in policing drug quality in foreign countries. When infractions in regulations or product quality are spotted, such as finding mold or contaminants in machinery, instead of restricting the import of the items in question until standards improve, the inspectors generally downgrade the issues to self correction status. This allows the companies to voluntarily correct the problem, without penalty. This process has encouraged corruption, using substandard ingredients and short cuts that make many drugs ineffective and potentially harmful.
Whistleblower Uncovers Corruption
The extent to which substandard generic drugs have been allowed to enter the US would probably have never been known if it were not for a whistleblower who eventually exposed the fraud and deception that occurs to mislead FDA inspectors. The whistleblower, Dinesh Thakur, was an engineer and executive at Ranbaxy Laboratories, Limited, the largest generic drug maker in India. At the time, Ranbaxy supplied the US with 52 million prescription drugs per year. Thakur was born in India but received his college education and initial professional training in the United States before taking a position at Ranbaxy.
Assuming he would see the same level of professionalism and adherence to regulations as he had been accustom to in the US, he was shocked by the gross disregard to proper drug manufacturing practices and extensive fraud to deceive FDA inspectors. Employees routinely falsified documents and altered test results to show their products as passing quality standards. Inspectors looking over the documents would never have known they were faked. Instead of destroying products that failed testing, they were shipped to poorly regulated countries. In fact, according to Thakur, nearly all of the drugs Ranbaxy shipped to places like Africa, Southeast Asia, and the Middle East were defective. Many of the drugs they shipped to the US and Europe were also substandard. In May 2013, Ranbaxy pleaded guilty to selling adulterated drugs in the US and paid the US government $500 million in penalties and fines.
In 2015, two years after the Ranbaxy incident, the FDA issued a warning letter to Dr. Reddy’s Laboratories, the second largest generic drug manufacturer in India, supplying medicines to US. The FDA said that the company destroyed records of failing tests and kept only those that showed their products passing.[ 6] In between the Ranbaxy and Reddy cases, over 40 other instances of data fraud citations against manufacturing facilities in India occurred.
Indian companies have demonstrated that they have competence and the technology to produce inexpensive, high-quality generics. Yet, with largely non-existent domestic regulators, and a commerce department that acts as the publicity department for Indian generics, they continue to falter after being singled out for consistent wrongdoing by western regulators.
The Ranbaxy case made FDA inspectors more aware of the problem. Indian generics are the cheapest in the world, but there is increasing concern among doctors that Indian products sometimes do not work. Dr. Harry Lever runs the hypertrophic cardiomyopathy group at the Cleveland Clinic in Ohio and perhaps like many physicians he simply tries to avoid Indian medicines. “I simply can’t trust them anymore,” says Dr. Lever. “I’ve seen many patients who haven’t responded properly to them, and although I’ve complained repeatedly to FDA, they do very little.”[7]
Unannounced Inspections
The primary reason why FDA inspectors did not identify the problems at Ranbaxy earlier was because of the agency’s advance notice policy. With advance notice company employees were given time to alter documents, clean the facilities, and destroy incrementing evidence. In mid-2013, Altaf Lal was appointed to be the new head of the FDA India office. In order to clean up the mess exposed by the Ranbaxy incident, Lal proposed a pilot program to make all inspections in India unannounced or of short notice. The program was inaugurated in December 2013. The results were instantaneous.
In January 2014, the FDA was planning an unannounced inspection at a plant in northern India on Monday. Suspecting that company officials may have been alerted, the inspectors arrived a day early. Expecting to see the plant quiet on a Sunday morning, instead they found it bustling with activity. Dozens of employees were hunched over documents, backdating them. On a desk they spotted a notebook listing the documents the workers needed to fabricate in anticipation of the inspectors’ arrival. Post-it notes were stuck to documents, noting what dates to change.
According to Katherine Eban, the author of the book A Bottle of Lies: The Inside Story to the Generic Drug Boom, the focus of many companies is not on producing perfect drugs, but on producing perfect data to fool inspectors. At one plant, inspectors went straight to the microbiology laboratory and found the paperwork for testing the sterility of the plant in perfect order: microbial limits testing, biological indicators, all the samples with perfect results. Yet most of the samples didn’t exist. The plant was testing almost nothing. The laboratory, it turned out, was a fake.
Eban explains that in the vast majority of unannounced inspections, investigators found conditions the plant managers had no time to fix: infestations of birds and insects, a pile of critical manufacturing records tossed in a trash bin, backed up toilets in an employee bathroom with urine puddling directly onto the floor near a sterile manufacturing area.
One inspector highlighted in Eban’s book, Peter Baker, performed foreign inspections for the FDA for six years. On one of his first trips to India, Baker visited a plant in Aurangabad run by the Indian company Wockhardt, which made about 110 generic-drug products for the American market. He had one week at the plant to ensure that it complied with the FDA’s good manufacturing practices. Generating and preserving data at each manufacturing step is crucial to following those regulations.
On his second day at the Wockhardt plant, Baker and a colleague caught an employee trying to smuggle out a garbage bag of documents. The documents led Baker to discover that the plant had knowingly sold into foreign markets vials of insulin containing metallic fragments. These had apparently come from a defective sterilizing machine. He learned that the company had been using the same defective equipment to make a sterile injectable cardiac drug for the American market. The willful deception there and at other plants so shocked him that he overhauled his inspection methods, with significant results. Two months after Baker’s Wockhardt inspection, the FDA banned the import of drugs from that plant into the United States, a potential $100 million loss in sales for the company.
Baker kept digging. Over the next five years, first in India and then in China, he uncovered fraud or deceptive practices in almost four-fifths of the drug plants he inspected. Some of the plants used hidden laboratories, secretly repeated tests and altered results to produce fake data that fundamentally misrepresented drug quality, then submitted that data to regulators.
During his 27 months in India, Baker found fraudulent or deceptive data in 29 of the 38 drug plants he inspected. In 2015, Baker moved to China, where he found similar data fraud and deception in 38 of the 48 drug plants he inspected. Shaken by what he uncovered in his work for the FDA, he told a colleague that if people knew how some of their drugs were manufactured overseas “then no one would take them.”
The FDA Has Failed
Because of the great demand for cheap generic drugs in the US, prior to the pilot program, which began in 2013, when problems were discovered inspectors only infrequently invoked the most serious finding “official action indicated” that would result in the recall or ban of a drug until the problem was corrected. Instead, they usually chose the lesser designation “voluntary action indicated” and allow the company to correct the problem on their own while continuing to ship drugs into the United States. There was no rush to make the correction as inspectors only visited once every nine years or so. There was plenty of time to fix the problem or if not fixed simply hide it from the next inspector.
Under the pilot program, the rate of inspections resulting in the FDA’s “official action indicated” increased dramatically. Before long, drugs from numerous plants in India had been banned from the United States market. Given these results, it seemed logical for the FDA to make unannounced inspections or short notice the norm around the world. But in July 2015, FDA officials decided to terminate the program and return to largely pre-announced inspections in India. When asked why, the agency declined to explain its reasoning and stated that “after evaluation of the pilot a decision was made to discontinue the pilot.” From 2013 to 2018, FDA officials downgraded the regulatory sanctions against more than 100 Indian plants, changing the designation of “official action indicated” to “voluntary action indicated.”
An excuse the FDA has given for relaxing the restrictions on drug imports is that Americans need affordable drugs and if bans are placed on them then consumers will not have access to the drugs they need. This reasoning seems illogical. Cheap drugs are not a bargain if they don’t work. If they cause harm they are worse than nothing at all.
Despite the changes, the FDA has reassured the public that they will continue to do unannounced inspections at foreign manufacturing facilities if they are warranted. The FDA insists that its import alerts, which can stop substandard drugs from entering the country, are an effective tool for protecting patients.
Can We Trust Domestic Drugs?
Drug-quality problems are not restricted to areas where oversight is weak. The FDA has found quality failures in domestic manufacturing as well. When a problem is found, drugs are often recalled to protect the public. The reasons for recalls include violation of GMP, lack of processing controls, microbial contamination, subpotent active ingredient, presence of particulate matter, defective delivery system, non-sterility, damage from improper storage conditions, the presence of foreign substance, and incorrect product formulation, among others.

We only occasionally hear of a drug recall. It is so infrequent that we tend to assume that recalls are a rare incidence, but that is not the case. Recalls occur all the time and in surprising numbers. Over a 10-year period from 1999-2009 there were over 4,500 pharmaceutical products recalled in the United States. In 2009 alone 1,384 drug products were recalled. [8] What if you are taking one or more of these drugs, how are you to know there is a problem with them? You likely won’t. We seldom hear about these, perhaps because they are a daily occurrence. In 2018 there were 702 drugs recalled, that equates to nearly two a day! Of that number, 689 were for products manufactured in the US, In contrast, the number of recalled drugs from China were only 2 and those from India were only 10 and their drug production is much less likely to follow the FDA’s GMP and produce a much greater number of substandard or defective products.[ 9] Which means a lot of substandard or potentially substandard drugs are currently being marketed in the US.
A report by the PEW Health Group published in 2011, mentions four examples of the hundreds of recalls that occurred in 2010. [10]
In 2010, Johnson & Johnson recalled more than 130 million bottles of children’s cough and cold medicines after an FDA inspection revealed 20 alleged GMP violations, including failure to conduct an adequate investigation of inactive ingredients that were contaminated with bacteria.
In April 2010, the FDA discovered ibuprofen tablets made by Michigan-based L. Perrigo Co were contaminated with metal shavings. The FDA has reported GMP and compliance issues at Perrigo since 2005. In 2006, that company recalled 384 lots of acetaminophen tablets after the discovery of metal particles in some of their drugs.
In May 2010, Genzyme Corp. agreed to pay $175 million after the FDA discovered serious manufacturing quality issues at the company’s Allston, MA, plant. Alleged violations included drug contamination with metal, glass, and rubber particles, and viral contamination in manufacturing equipment.
In October 2010, GlaxoSmithKline paid a penalty of $750 million for distributing adulterated drugs that did not meet necessary levels of strength, purity, or quality. Some of the drugs contained the wrong amount of active ingredient, and some were possibly exposed to contamination by microorganisms.
Over the past several years much of the US drug manufacturing has moved overseas, principally India and China. With less oversight, manufacturers may not rigorously observe quality measures, and in some cases individuals manage to deliberately substitute cheaper materials for high-quality ingredients. While the vast majority of drugs in the United States are safe, these changes create significant risks of potentially serious adverse effects by which US patients are harmed by substandard or adulterated drugs. Even brand-name drugs are now being produced overseas with potential risks. A good example of the potential danger of outsourcing drug manufacturing to less well regulated countries occurred with heparin, the most commonly used blood thinner in the US.
In early 2007, the US Center for Disease Control and Prevention (CDC) and the FDA began receiving reports of unexpected allergic-like reactions and high blood pressure in patients undergoing dialysis. Reported events sharply increased in December 2007 and into 2008. Over the course of 18 months at least 178 confirmed deaths were reported along with hundreds of patients experiencing adverse reactions severe enough to be reported to government health officials. The events were subsequently linked to heparin, made by Baxter International, Inc. Additional analysis led to the identification of a chemical adulterant in the drug known as oversulfated chondroitin sulfate (OSCS), which had escaped detection on standard tests.
Baxter and 15 other companies in the US recalled at least 11 heparin drugs and 72 medical device products (heparin is used to coat stents and in other medical devices). Heparin products were also recalled in Europe, Australia, and Japan. Once the drug was removed from the market, the unusual adverse reactions ceased.
It was later determined that the adulteration of heparin occurred during manufacture in China. Heparin is derived from animal mucosal tissues, almost exclusively from pigs. In China, numerous workshops harvest basic heparin material (heparin crude) by cooking and drying pig intestines collected from local slaughterhouses. These workshops are often run by independent business owners and are subject to limited scrutiny. Heparin crude is then sold, often through consolidators, to other plants that further refine the material into the active ingredient and then combine it with fillers or inactive ingredients to produce the final product.
OSCS is a synthetic chemical that costs nearly 100 times less to produce than actual heparin. Because it mimics some of heparin’s chemical properties, it was not detected as an adulterant by standard assays. Somewhere along the manufacturing process the cheaper OSCS was substituted to cut costs and increase profits. The difficulty of distinguishing the adulterant from the active ingredient made the deception hard to detect; if it were not for the severe adverse reactions caused by the adulterant, the deception may not have been discovered for years. Even if the adulterant caused no harmful reactions itself, using it in place of the active ingredient would have resulted in millions of patients receiving drugs that were ineffective or of reduced potency, greatly increasing the risk of complications and death.
In 2008, when Baxter sent inspectors to China to examine the plants and evaluate the supply chain, they were denied access to certain facilities processing heparin. FDA inspectors were also denied access. The investigators, however, did discover a number of manufacturing quality issues, including poor control of incoming raw materials. The FDA was unsuccessful in getting cooperation from Chinese authorities to investigate further. We can assume that denying inspectors access to certain facilities was done to avoid discovering gross problems in the manufacturing process that could seriously affect drug quality and purity. Unfortunately, the FDA took no action against the companies involved as the investigators could not confirm exactly where in the manufacturing process the adulteration occurred. Consequently, the manufacturers are still supplying drugs, which may have quality issues, to the world market.
What You Can Do
If we knew where our drugs were manufactured, we could make an educated decision whether or not to use them, weighing the risk against the benefits. But we don’t always have that option. Unlike most other consumer products, drug labels may not offer consumers a clear picture of the countries in which they were manufactured. US statute requires all imported products to display to the end customer their country of origin. Imported finished drugs therefore, must list the country where the manufacturing occurred. But if a company imports pharmaceutical ingredients and then formulates those substances into a medicine in the United States, the form of the product has changed, and the country of origin labeling requirements don’t apply. If the manufacturer is named, it is the maker of the finished product and not necessarily the maker of the active ingredient.
Foreign drug makers have proven that they have the technical knowledge, ability, and equipment to produce high-quality pharmaceuticals. Most of the generic and brand-name drugs in the US are exactly what they are supposed to be. You take a greater risk with imported generics. Even if only 10 percent of them are substandard or adulterated that equates to millions of drugs. Since there are risks to taking any drug, generic or not, it is best to use medicines only when they are absolutely needed. At times, the brand-name drug may be your best choice. And if they don’t work or cause unwanted reactions, don’t use them.
References:
1. US Government Accountability Office (September 2010). Drug Safety: FDA Has Conducted More Foreign Inspections and Begun to Improve Its Information on Foreign Establishments, but More Progress is Needed. Appendix 1: Comments from the Department of Health and Human Services (Publication No. GAO-10-961).
2. US Government Accountability Office (March 1998). Food and Drug Administration: Improvements Needed in the Foreign Drug Inspection Program (Publication No. GAO/HEHS-98-21).
3. International Medical Products Anti-Counterfeiting Taskforce, World Health Organization. “Counterfeit Drugs Kill!” Updated May 2008.
4. Mason, RP, et al. Atorvastatin generics obtained from multiple sources worldwide contain a methylated impurity that reduces their HMG-CoA reductase inhibitory effects. Journal of Clinical Lipidology 2013;7:287.
5. Eban, K. Bottle of Lies: The Inside Story of the Generic Drug Boom. Ecco Press: New York, 2019.
6. [ https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/warning-letters/dr-reddys-laboratories-limited-11052015
7. https://www.realclearhealth.com/articles/2016/06/01/indias_faulty_medicine_and_the_risk_to_patients_109908.html
8. Cox, B. Record Drug Recall Totals for 2009 Resulted from GMP Breakdowns. The Gold Sheet, May 2010, Vol. 44, No.5.
9. https://www.pharmatimesnow.com/2019/02/top-20-reasons-for-drug-recalls-in-usa.html
10. PEW Health Group. After Heparin: Protecting Consumers from the Risks of Substandard and Counterfeit Drugs. Washington, DC: 2011.

Author, Katherine Eban, argues that generic drugs are poisoning us. “My reporting on the generic drug industry over the last decade led me to four continents, and into the overseas plants where America’s generic drugs are made,” says Eban. “Interviews with more than 240 people, including numerous whistleblowers, helped expose what was going on behind the boardroom doors at generic drug companies. Some companies have encouraged data fraud as the most profitable path to securing approvals from regulators, and have used deceit to hold the FDA’s investigators at bay.”
(Paid Link)
Bottle of Lies is an invaluable exposé of the generic drug industry. It reads like a mystery novel uncovering the clues from investigators and whistleblowers that exposes the entire foreign generic drug trade.
You can learn more about this book on Amazon here.

Plastics contain a number of chemicals that pose a threat to our health. The most notorious perhaps are Bisphenol-A (BPA) and phthalates. These chemicals leach into food and beverages from plastic containers and wraps and ultimately collect in your body. BPA is particularly worrisome because it is so common. BPA is an estrogen mimic and can disrupt normal hormone function and sexual development. Other health risks include brain damage and psychological disorders, increased fat accumulation, altered immune function, stimulation of prostate size, liver disease, diabetes, and heart disease. Phthalates are a group of chemicals used to make plastics more flexible and resilient. They are potent endocrine disrupters.
These chemicals are found in plastic water bottles and milk jugs, microwavable plates and utensils, canned foods and soda cans (most have plastic lining in the cans), baby bottles and pacifiers, and nearly all processed food packaging, including the plastic used to wrap fresh meats. These chemicals leach into our foods over time. If the food is hot or heated in an oven or microwave the rate of absorption dramatically increases.
Another source of plastic comes from teabags. Most “paper” teabags are infused with plastic. When the teabag is seeping in hot water, microparticles of plastic from the bag are released into your tea. The degree of plastic pollution released from teabags was uncovered by researchers from McGill University. The researchers used four commercial products. The tea leaves were removed to ensure that none of the plastic came from the leaves themselves. The empty bags were placed in hot water to simulate the seeping process. The researchers found that a single bag released billions of plastic particles into the hot water. They reported that the level of plastic released was thousands of times greater than has been reported with other foods and beverages.
Most teabags, and even coffee filters, are treated with plastic to reduce the chances of tearing during use. When you look at a teabag it doesn’t seem like it would contain plastic, but most teabags are composed of about 25 percent plastic.
To learn more about the plastic in teabags and how to avoid it, you can read the full story here

Issue 16-5 covers: Is Coconut Oil Toxigenic?—Protect Your Vision with Coconut Oil—Natural Versus Allopathic Medicine—Coconut Oil for A Healthy Planet
